Protocolo testado utilizando o Linux Ubuntu 24 LTS
Os passos iniciais são utilizados para instalação dos softwares necessários, preparação da estrutura de diretórios e obtenção dos arquivos utilizados.
1)Instalação do programa HiSat2:
No terminal do linux, utilize o comando:
sudo apt-get install hisat2

*a versão instalada foi 2.2.1
2)Instalação do programa samtools:
*Version: 1.19.2
no terminal digite:
sudo apt update
sudo apt install samtools
2.1)caso queira remover uma instalação antiga, digite:
sudo apt remove samtools
sudo apt autoclean && sudo apt autoremove
3)Instalação do programa HTseq-count:
consulte:
https://shicheng-guo.github.io/research/1941/01/08/HTseq
4)Criar estrutura de diretórios:
4.1) criar um diretório chamado rna1:
mkdir rna1
entrar no diretório rna1:
cd rna1
4.2) criar sub-diretórios:
mkdir fastq
mkdir ref
mkdir bam

5) Fazer download do arquivos do genoma de referencia (em formato fasta) e de anotação dos genes (formato gtf ou gff3):
*neste exemplo, serão utilizados dados do genoma do arroz, armazenados nos links:
https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_genome.fasta.gz – para arquivo das pseudo-moléculas do genoma do arroz
https://rapdb.dna.affrc.go.jp/download/archive/irgsp1/IRGSP-1.0_representative_transcript_exon_2024-07-12.gtf.gz – para arquivo formato gtf, contendo anotação das coordenadas dos genes em cada cromossomo
**após o download, copie estes arquivos no diretório “ref”, contendo as informações do genoma de referência.
6) Fazer download dos arquivos contendo o sequenciamento dos RNAs, em formato fastq:
*existêm dois tipos de sequenciamento:
SE (single end), onde somente um lado dos fragmentos são sequenciados.
PE (paired end), onde os dois lados dos fragmentos são sequenciados. Neste caso, os dados de cada amostra sequenciada sao representados por dois arquivos, contendo as reads (sequencias) forward e reverse, separadamente.
6.1)baixar os arquivos para o diretório fastq;
link para arquivos:
https://docs.ufpel.edu.br/index.php/s/6nDhkMF4ChgULFz
sample1_1.fastq.gz
sample1_2.fastq.gz
sample2_1.fastq.gz
sample2_2.fastq.gz
sample3_1.fastq.gz
sample3_2.fastq.gz
sample4_1.fastq.gz
sample4_2.fastq.gz
sample5_1.fastq.gz
sample5_2.fastq.gz
sample6_1.fastq.gz
sample6_2.fastq.gz
* É importante informar que estes dados contem somente reads pertencentes ao cromossomo 1 do arroz, derivados o trabalho de Amaral et al., (2016) https://link.springer.com/article/10.1007/s10142-016-0507-y.
***Estes dados representam uma parcela muito pequena do que realmente é o transcriptoma do arroz, por isso as conclusões obtidas neste exemplo são somente didáticas.
***estes dados foram reduzidos somente ao cromossomo 1 para facilitar a execução didática dos métodos.
================================================================================================
***Os próximos passos, são utilizados para a análise de fato.
7)Criação do arquivo de índice para o genoma de referência:
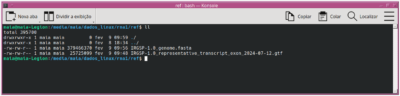
*na pasta “ref”, temos que descompactar os arquivos baixados.
7.1)comandos para descompactar arquivos:
gunzip IRGSP-1.0_genome.fasta.gz
gunzip IRGSP-1.0_representative_transcript_exon_2024-07-12.gtf.gz
resultado:
*posteriormente, é aconselhável re-nomear os arquivivos para nomes mais simples/curtos, exemplo:
IRGSP-1.0_genome.fasta
para
genome.fasta
IRGSP-1.0_representative_transcript_exon_2024-07-12.gtf
para
genes.gtf
para isso, use o comando move (mv):
mv IRGSP-1.0_genome.fasta genome.fasta
mv IRGSP-1.0_representative_transcript_exon_2024-07-12.gtf genes.gtf
resultado: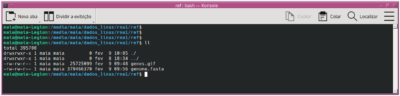
7.2) comando para criar índice:
hisat2-build -p 15 genome.fasta genome_index
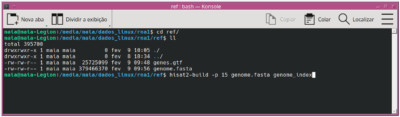
resultado:
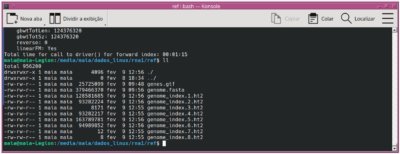
8) Renomear as amostras (arquivos fastq):
*para facilitar o trabalho, é importante renomear os arquivos fastq de acordo com nomes que facilitem a analise:
**neste exemplo, o experimento é composto de três repetições (amostras) de uma cultivar de arroz nas condições de estresse por ferro e sal, então, vamos nomear cada arquivo conforme as informações originais do experimento.
***também podemos facilitar a compreensão indicando nos nomes quem é a amostra sequenciada no sentido forward e reverse.
****por padrão, arquivos com reads forward, recebem o numero 1 e reverse o numero 2:
exemplo:
sample1_1.fastq.gz, equivale a: sample1_forward.fastq.gz ou sample1_f.fastq.gz
sample1_2.fastq.gz, equivale a: sample1_reverse.fastq.gz ou sample1_r.fastq.gz
*****utilizaremos, somente f e r, para simplificar.
para este exemplo, as informações das amostras estão no arquivo
https://docs.ufpel.edu.br/index.php/s/M30IxguzmuL1F9o

então, utilizando o comando mv, podemos renomear os arquivos para:
sample1_1.fastq.gz – frio_r1_f.fastq.gz
sample1_2.fastq.gz – frio_r1_r.fastq.gz
sample2_1.fastq.gz – frio_r2_f.fastq.gz
sample2_2.fastq.gz – frio_r2_r.fastq.gz
sample3_1.fastq.gz – frio_r3_f.fastq.gz
sample3_2.fastq.gz – frio_r3_r.fastq.gz
——-
sample4_1.fastq.gz – sal_r1_f.fastq.gz
sample4_2.fastq.gz – sal_r1_r.fastq.gz
sample5_1.fastq.gz – sal_r2_f.fastq.gz
sample5_2.fastq.gz – sal_r2_r.fastq.gz
sample6_1.fastq.gz – sal_r3_f.fastq.gz
sample6_2.fastq.gz – sal_r3_r.fastq.gz
resultado:
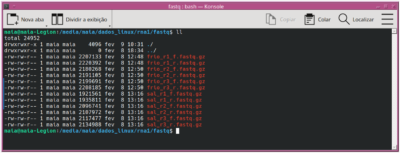
9) Mapear as amostras (arquivos fastq) contra o genoma de referência (indexado):
*para isso permaneça no diretório “rna1”;
**certifique-se que foi criada a pasta “bam”;
***o comando utilizado ira ler os arquivos de reads da pasta “fastq”, o genoma de referencia da pasta “ref” e gravar as saidas na para “bam”;
no terminal digite:

para amostra 1:
hisat2 –rna-strandness RF –very-sensitive -k 5 -p 15 –dta -x ref/genome_index
-1 fastq/frio_r1_f.fastq.gz -2 fastq/frio_r1_r.fastq.gz -S bam/frio1.sam –summary-file frio1_summary.txt
resultado:
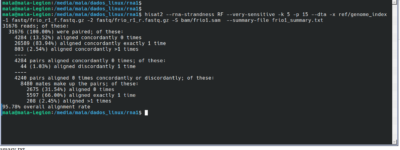
para amostra 2:
hisat2 –rna-strandness RF –very-sensitive -k 5 -p 15 –dta -x ref/genome_index
-1 fastq/frio_r2_f.fastq.gz -2 fastq/frio_r2_r.fastq.gz -S bam/frio2.sam –summary-file frio2_summary.txt
****repetir para todas as amostras, alterando os nomes dos arquivos de entrada e saída adequadamente
resultados:
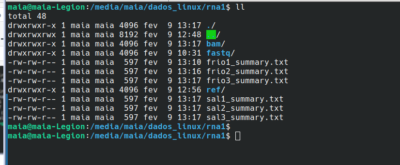
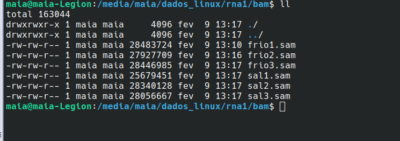
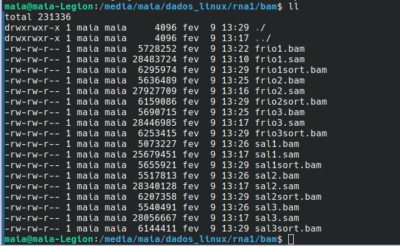
10) Converter os arquivos formato “sam” para formato “bam” e ordená-los:
10.1) converter sam para bam:
entre na pasta bam e execute o comando:
samtools view -@ 15 -S -b frio1.sam > frio1.bam
samtools view -@ 15 -S -b frio2.sam > frio2.bam
**repetir o comando para todas as amostras.
10.2) ordenar arquivos “bam”:
execute o comando:
samtools sort -@ 15 -O BAM -o frio1sort.bam frio1.bam
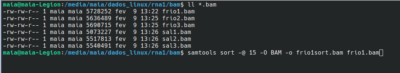
**repetir o comando para todas as amostras.
resultado:
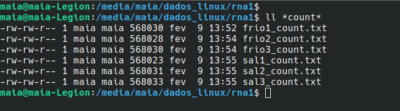
11) Contagem de reads mapeados nos genes anotados:
vá para a pasta “rna1” e execute o comando:
para amostra 1:
htseq-count –stranded=reverse -f bam bam/frio1sort.bam ref/genes.gtf > frio1_count.txt
para amostra 2:
htseq-count –stranded=reverse -f bam bam/frio2sort.bam ref/genes.gtf > frio2_count.txt
**repetir o comando para todas as amostras.
resultado:
**cada arquivo de contagem vai ter na primeira coluna a identificação dos genes e a segunda coluna contendo o total de reads mapeados dentro daquele gene, conforme figura abaixo.

12) Montagem de planilha do excel juntando os resultados de contagem de todas as amostras:
** a partir desta ponto, as demais atividades podem ser feitas utilizando o Office e R no sistema MS-Windows.
12.1) arquivo de contagens
exemplo:
dados da amostra frio1_count.txt
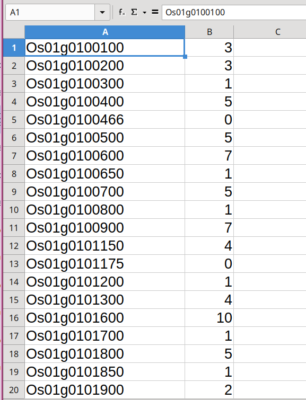
dados da amostra frio2_count.txt

**unificando as planilhas:
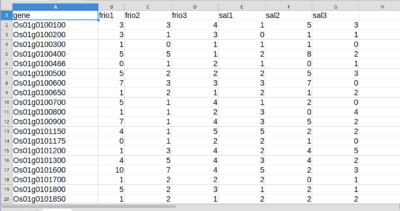
*no excel, a primeira coluna devera receber os nomes dos genes e as demais, os nomes de cada amostra.
**na primeira linha deverão ser colocados os nomes de cada coluna.
a primeira devera ser nomeada como “gene” e as demais com o nome dos tratamentos e repetição, conforme exemplo acima.
***é importante manter padrão na escrita, evitando espaços, acentos, caracteres especiais e manter apenas letras minúsculas, para simplificar.
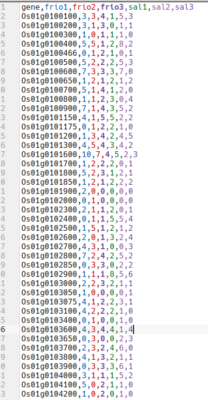
****os dados do excel deverão ser exportados para arquivo formato csv (contagem_final.csv), conforme exemplo abaixo:
contagem_final.csv
12.2) arquivo com desenho experimental
no excel, monte um arquivo com a estrutura conforme a figura abaixo:
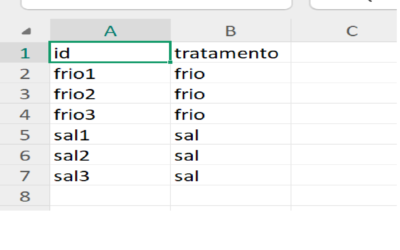
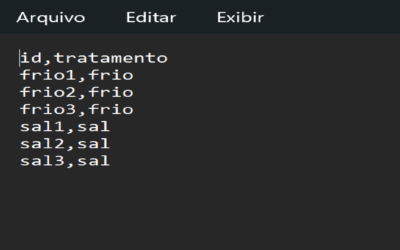
exporte esses dados para um arquivo csv chamado “metadata.csv”, por exemplo.
13) Instalação do R e demais pacotes:
** a partir desta ponto, as demais atividades podem ser feitas utilizando o Office e R no sistema MS-Windows.
a) Inicialmente necessitamos instalar o R e RStudio;
*neste exemplo foi utilizado o R versão 4.4.0, obtido em:
https://cran.r-project.org/bin/windows/base/old/
ou
https://cran.r-project.org/bin/windows/base/old/4.4.0/
b) Instalação do DESeq2:
Inicialmente é necessário instalar o gerenciador de pacotes, com os comandos:
if (!require(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
c) Instalar o DESeq:
BiocManager::install(“DESeq2”)
14) Análise da expressão diferencial com o pacote DESeq2 no RStudio:
*o protocolo descrito é derivado (com algumas simplificações) do artigo:
Count-based differential expression analysis of RNA sequencing data using R and Bioconductor (Anders et al., 2013 – Nature Protocols)
https://www.nature.com/articles/nprot.2013.099
14.1)leitura dos dados
#ler bibliotecas
library( “DESeq2” )
library(“ggplot2”)
#define pasta
setwd(“e:/rna1/”)
getwd()
#ler dados de contagem
countdata <- read.csv(“contagem_final.csv”, header = TRUE, sep = “,”)
head(countdata)
#ler dados da estrutura do experimento
exp_design <- read.csv(“metadata.csv”, header = TRUE, sep = “,”)
exp_design
#indicar que a coluna tratamento é um fator
exp_design$tratamento = factor(exp_design$tratamento)
#é indicado fazer uma limpeza prévia, excluindo genes com valor baixo de expressão ou zero, nesse caso será utilizado um mínimo de 1 reads em todas a três amostras do frio OU do sal
para ou utiliza-se o caractere pipe (“|”)
minimo = 1
countdata2 <- subset(countdata, (frio1 >= minimo & frio2 >= minimo & frio3 >= minimo) | (sal1 >= minimo & sal2 >= minimo & sal3 >= minimo))
dim(countdata2)
#criar dataset do deseq2
dds <- DESeqDataSetFromMatrix( countData=countdata2 ,
colData=exp_design,
design=~tratamento, tidy = TRUE)
dds
dim(dds)
resultado no console do R:
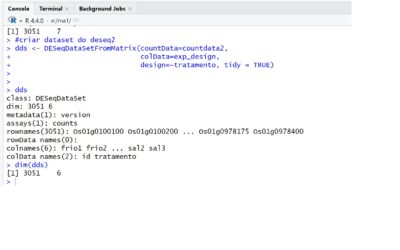
# filtragem 2
smallestGroupSize <- 3
valor <- 1
keep <- rowSums(counts(dds) >= valor) >= smallestGroupSize
dds <- dds[keep,]
dds
dim(dds)
#indicar qual nível do fator é referencia no calculo de expressao diferencial
dds$tratamento <- relevel(dds$tratamento, ref = “sal”)
#executar a funcao DESeq para calculo da diferenca de expressao
dds <- DESeq(dds)
res <- results(dds)
res
resultado console do R:

#sumário dos resultados
summary(res)
#exemplo de como selecionar somente os significativos
res0.05 <- results(dds, alpha = 0.05)
summary(res0.05)
#nomes do contraste feito
resultsNames(dds)
resultado:
“tratamento_frio_vs_sal”
#criar manualmente um contraste – é importante quando o fator tratamento tem vários níveis, exemplo frio, sal, calor, dose1, dose2
contrast <- results(dds, name=”tratamento_frio_vs_sal”)
contrast <- results(dds, contrast=c(“tratamento”,”frio”,”sal”))
contrast
#o shrinkage” (ou encolhimento) dos valores de log2 da razão de expressão (logfold changes) é uma técnica usada no deseq2 para melhorar os valores da expressão diferencial
#deve ser utilizada em situações onde há poucos dados ou com muita variabilidade
#o apeglm é um método bayesiano aplicado nestas condições
#existem outros métodos, mas protocolo dos autores do deseq2 ele indica este método
resLFC <- lfcShrink(dds, coef=”tratamento_frio_vs_sal”, type=”apeglm”)
resLFC
#plotar gráficos de dispersao dos resultados de log2fc
plotMA(res, ylim=c(-2,2))
plotMA(resLFC, ylim=c(-2,2))
#ajustar os dados
#Variance Stabilizing Transformation (vst) transforma dos dados de contagem
#para quye a variabilidade entre as amostras seja estabilizada
#ele “homogeniza as variância”
#rlog (regularized log transformation) é outro metodo com a mesma finalidade
#blind=FALSE utiliza informações das condicoes do experimento para os calculos
#normTransform – para reduzir outliers
# todas sao transformações
#neste exemplo vamos utilizar os dados transformados via rlog
#conforme artigo https://doi.org/10.1186/s13059-014-0550-8
vsd <- vst(dds, blind=FALSE)
rld <- rlog(dds, blind=FALSE)
ntd <- normTransform(dds)
head(assay(vsd), 3)
meanSdPlot(assay(ntd))
meanSdPlot(assay(vsd))
meanSdPlot(assay(rld))
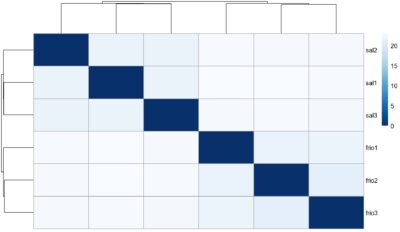
# plotar gráfico de distancia de PCA entre as amostras
sampleDists <- dist(t(assay(rld)))
sampleDistMatrix <- as.matrix(sampleDists)
rownames(sampleDistMatrix) <- paste(rld$id)
colnames(sampleDistMatrix) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, “Blues”)) )(255)
pheatmap(sampleDistMatrix,
clustering_distance_rows=sampleDists,
clustering_distance_cols=sampleDists,
col=colors)
plotPCA(rld, intgroup=c(“id”))
plotPCA(rld, intgroup=c(“tratamento”))
resultados:

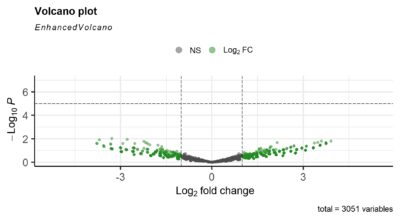
#https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html
#gráfico v0lcano – neste caso como a amostra
EnhancedVolcano(res,lab = ”, x = ‘log2FoldChange’, y = ‘pvalue’)
EnhancedVolcano(res,lab = ”, x = ‘log2FoldChange’, y = ‘pvalue’) + coord_flip()
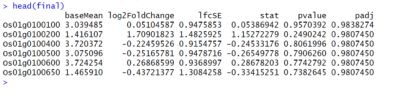
criar dataframe contendo os DEGs
final <- as.data.frame(res)
head(final)
#boxplot das amostras
par(mar=c(8,5,2,2))
boxplot(log10(assays(dds)[[“cooks”]]), range=0, las=2)
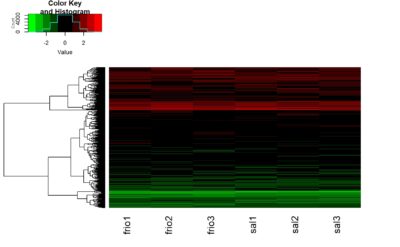
##criar heatmaps
library(gplots)
dados1 <- assay(rld)
#qdo quiser selecionar somente as primeiras 1000 linhas ou outro valor
#dados1 <- dados1[1:10000,]
#aqui utilizaremos todos os dados
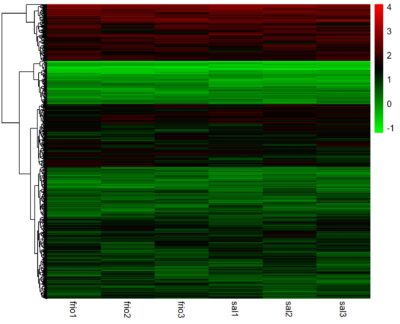
pheatmap(as.matrix(dados1),
color=greenred(75),
show_rownames = FALSE,
cluster_rows=TRUE,
cluster_cols=FALSE)
resultado:
pheatmap(as.matrix(dados1),
show_rownames = FALSE,
cluster_cols=FALSE,
colorRampPalette(c(“darkgreen”, “black”, “darkred”))(75),
border_color = “black”, # default is grey60
number_color = “black”,
fontsize_number = 8
)
pheatmap(as.matrix(dados1),
show_rownames = FALSE,
cluster_cols=FALSE,
colorRampPalette(c(“darkgreen”, “white”, “darkred”))(75),
border_color = “black”, # default is grey60
number_color = “black”,
fontsize_number = 8
)

#mais modelos de heatmaps
anotacao <- data.frame(exp_design$tratamento)
row.names(anotacao) <- colnames(dados1)
colnames(anotacao)[1] =”tratamento”
anotacao
cores = list(
tratamento = c( frio = “blue”,
sal = “magenta”)
)
pheatmap(as.matrix(dados1),
show_rownames = FALSE,
cluster_cols=FALSE,
colorRampPalette(c(“darkgreen”, “white”, “darkred”))(75),
border_color = “black”,
number_color = “black”,
fontsize_number = 8,
annotation_col = anotacao)
pheatmap(as.matrix(dados1),
show_rownames = FALSE,
cluster_cols=FALSE,
colorRampPalette(c(“darkgreen”, “white”, “darkred”))(75),
border_color = “black”,
number_color = “black”,
fontsize_number = 8,
annotation_col = anotacao,
annotation_colors = cores)
pheatmap(dados1,
cluster_rows = TRUE,
cluster_cols = TRUE,
clustering_distance_cols = ‘euclidean’,
clustering_distance_rows = ‘euclidean’,
clustering_method = ‘ward.D’,
annotation_names_row = FALSE,
annotation_names_col = TRUE,
fontsize_row = 10, # fonte das linhas
fontsize_col = 14, # fonte das colunas
angle_col = 45, # anglo das caixas de texto
show_colnames = T,
show_rownames = F,
colorRampPalette(c(“darkgreen”, “white”, “darkred”))(75),
annotation_col = anotacao,
annotation_colors = cores,
cutree_cols = 2
)
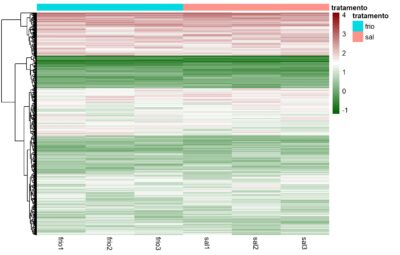

#centralizando (normalizando) os dados no heatmap
normalizado <- as.data.frame(scale(dados1))
pheatmap(normalizado,
cluster_rows = TRUE,
cluster_cols = TRUE,
clustering_distance_cols = ‘euclidean’,
clustering_distance_rows = ‘euclidean’,
clustering_method = ‘ward.D’,
annotation_names_row = FALSE,
annotation_names_col = TRUE,
fontsize_row = 10, # fonte das linhas
fontsize_col = 14, # fonte das colunas
angle_col = 45, # anglo das caixas de texto
show_colnames = T,
show_rownames = F,
colorRampPalette(c(“darkgreen”, “white”, “darkred”))(75),
annotation_col = anotacao,
annotation_colors = cores,
cutree_cols = 2
)
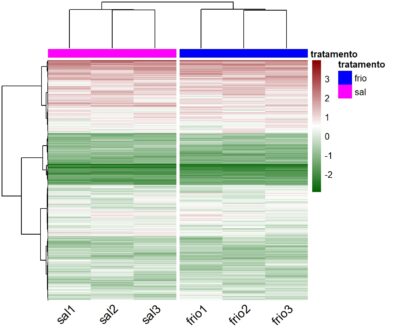
#dados2 <- dados1[1:5000,]
dados2 <- dados1
normalizado <- as.data.frame(scale(dados2))
heatmap.2(dados2,
key=T,
keysize=1.0,
scale=”none”,
density.info=”none”,
reorderfun=function(d,w) reorder(d, w, agglo.FUN=mean),
trace=”none”,
cexRow=0.2,
cexCol=0.8,
distfun=function(x) dist(x, method=”euclidean”),
hclustfun=function(x) hclust(x, method=”ward.D2″))
heatmap.2(as.matrix(normalizado),
dendrogram=”row”,
trace=”none”,
labRow=FALSE,
col=greenred(10))
heatmap.2(as.matrix(normalizado),
dendrogram=”row”,
trace=”none”,
labRow=FALSE,
col=redblue(256))
n <- nrow(normalizado)
color <- colorRampPalette((c(“blue”, “white”, “red”)))(n)
heatmap.2(as.matrix(normalizado),
dendrogram=”row”,
trace=”none”,
labRow=FALSE,
col=color)
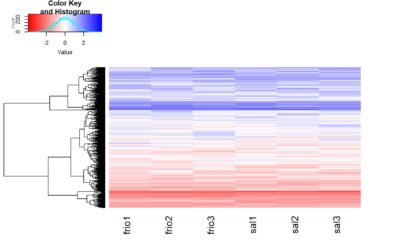
#correlação entre as amostras
dados3 <- dados1
dados3[is.na(dados3)] <- 0
cormat <- round( cor(dados3, method = c(“pearson”)), 2)
cormat
library(reshape2)
melted_cormat <- melt(cormat)
head(melted_cormat)
#Get the lower and upper triangles of the correlation matrix
#Get lower triangle of the correlation matrix
lower_tri <- cormat
lower_tri[lower.tri(lower_tri)] <- NA #OR upper.tri function
lower_tri
melted_cormat <- reshape2::melt(lower_tri, na.rm = TRUE)
# Heatmap
ggplot(data = melted_cormat, aes(Var2, Var1, fill = value))+
geom_tile(color = “white”)+
scale_fill_gradient2(low = “blue”, high = “red”, mid = “white”,
midpoint = .50, limit = c(0,1), space = “Lab”,
name=”Pearson\nCorrelation”) +
theme_minimal()+
theme(axis.text.x = element_text(angle = 45, vjust = 1,
size = 12, hjust = 1))+
coord_fixed() +
geom_text(aes(Var2, Var1, label = value), color = “black”, size = 4) +
theme(
axis.title.x = element_blank(),
axis.title.y = element_blank(),
panel.grid.major = element_blank(),
panel.border = element_blank(),
panel.background = element_blank(),
axis.ticks = element_blank(),
legend.justification = c(1, 0),
legend.position = c(0.4, 0.7),
legend.direction = “horizontal”)+
guides(fill = guide_colorbar(barwidth = 5, barheight = 1,
title.position = “top”, title.hjust = 0.5))

##distancia – dendograma entre amostras
t5 <- t(dados3)
distx = dist(t5, method = “manhattan” )
distx <- log10(distx)
hclust_avg <- hclust(distx , method = ‘average’)
plot(hclust_avg)

##mds entre amostras
fit <- cmdscale(distx, eig = TRUE, k = 2)
xx <- data.frame(fit$points[,1])
yy <- data.frame(fit$points[,2])
nome <- data.frame(row.names(t5))
nome
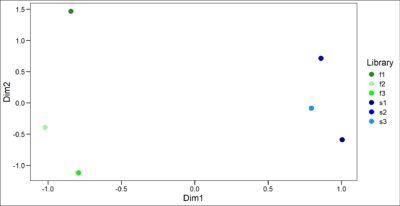
lin <- c(‘f1′,’f2’, ‘f3’,
‘s1′,’s2’, ‘s3’)
mds1 <- cbind(lin,xx)
mds1 <- cbind(mds1,yy)
colnames(mds1)[1] <- ‘Library’
colnames(mds1)[2] <- ‘Dim1’
colnames(mds1)[3] <- ‘Dim2’
library(ggthemes)
ggplot() +
geom_point(data = mds1, aes(x = Dim1, y = Dim2, color = Library),size=4) +
scale_color_manual(values = c(“forestgreen”, “palegreen”, “green”,
“darkblue”, “blue3”, “dodgerblue”)) +
theme_base()